A Fortran code to simulation the non-adiabatic Molecular Dynamics in the solid We using a linear vibroninc coupling model implementation of Tully's Fewest Sw a propagator and an implementation of Tully's model problems described in Tull
The code are writen in Fortran, and need MKL Fortran 95 library. To compiler t
module load compiler/intel/2020.4.302
tar -zxvf lvcsh.tar.gz
cd LVCSH
cd make
make
cd ../make_complex
make The the code use the EPW output file as main input file. Now only the version epw-code.org/) source code and recompile to print out the gmnvkq and vmef in c
cp LVCSH/docs/QE_change_code/v6.8/* qe-6.8/EPW/src
cd qe-6.8
make epw-
make a work directory (Carbyne)
-
In work directory do epw calculate, to get the electron-phonon coupling mat
(work directory) ___________________|_______________________________ | | | | | | | relax pseudo scf dos phonon epw LVCSH计算电声耦合项的步骤如下所示。在不同的目录下面进行结构优化、自洽计算、声子
2.1 进入relax目录,创建[vc-relax.in](http://www.quantum-espresso.org/Doc/IN [
forc_conv_thr](http://www.quantum-espresso.org/Doc/INPUT_PW.html#for quantum-espresso.org/Doc/INPUT_PW.html#press_conv_thr)的精度需要设置的比较vc-relax.in &CONTROL calculation = "vc-relax" restart_mode = "from_scratch" prefix = "carbyne" outdir = "./" pseudo_dir = "./pseudo/" verbosity = "high" tprnfor = .true. tstress = .true. etot_conv_thr = 1.0d-5 forc_conv_thr = 1.0d-4 / &SYSTEM ibrav = 0 nat = 2 ntyp = 1 !nbnd = 16 occupations = 'fixed' ! occupations = "smearing" ! smearing = "cold" ! degauss = 1.0d-2 ecutwfc = 50 ecutrho = 400 / &ELECTRONS conv_thr = 1.000e-7 electron_maxstep = 200 mixing_beta = 7.00000e-01 startingpot = "atomic" startingwfc = "atomic+random" / &IONS ion_dynamics = "bfgs" / &CELL cell_dofree = "x" cell_dynamics = "bfgs" press_conv_thr = 0.02 / K_POINTS {automatic} 200 1 1 0 0 0 ATOMIC_SPECIES C 12.01070 C.pbe-n-kjpaw_psl.1.0.0.UPF CELL_PARAMETERS (angstrom) 2.565602620 0.000000000 0.000000000 0.000000000 10.000000000 0.000000000 0.000000000 0.000000000 10.000000000 ATOMIC_POSITIONS (angstrom) C 0.0002913894 5.0000000000 5.0000000000 C 1.2644833916 5.0000000000 5.0000000000
采用下面命令查看计算过程中的受力情况已经压力张量和晶格常数变化情况
cat vc-relax.out |grep -A 12 "Total force ="
计算结束后,使用下面命令得到优化后的结果
grep -B 30 "Begin final coordinates" vc-relax.outawk '/Begin final coordinates/,/End final coordinates/{print $0}' vc-relaxBegin final coordinates new unit-cell volume = 1731.62982 a.u.^3 ( 256.60106 Ang^3 ) density = 0.15545 g/cm^3 CELL_PARAMETERS (angstrom) 2.566010647 0.000000000 0.000000000 0.000000000 10.000000000 0.000000000 0.000000000 0.000000000 10.000000000 ATOMIC_POSITIONS (angstrom) C 0.0019333750 5.0000000000 5.0000000000 C 1.2630425527 5.0000000000 5.0000000000 End final coordinates2.2
cp vc-relax.in relax.in将上面优化得到的[CELL_PARAMETERS](https html#ATOMIC_POSITIONS) 结果在relax.in中进行修改,并修改为`calculation = "recat relax.out |grep -A 12 "Total force ="
grep -B 30 "Begin final coordinates" vc-relax.outawk '/Begin final coordinates/,/End final coordinates/{print $0}' relax.ou得到如下结果:
Begin final coordinates ATOMIC_POSITIONS (angstrom) C 0.0019333750 5.0000000000 5.0000000000 C 1.2630425527 5.0000000000 5.0000000000 End final coordinates
2.3
cp relax.in scf.in将上面的原子位置(ATOMIC_POSITIONS)结果在scf.outdir= './'进行自洽计算。2.4 计算态密度.修改
calculaiton='nscf'并增加kpoint的采样密度。cp -r scf dos cd dos mv scf.in nscf.in使用dos.x对nscf计算得
&DOS prefix = 'carbyne' outdir = './outdir' bz_sum = "smearing" ngauss = 0 degauss = 2.0d-2 DeltaE = 0.01 fildos = 'carbyne.dos' /
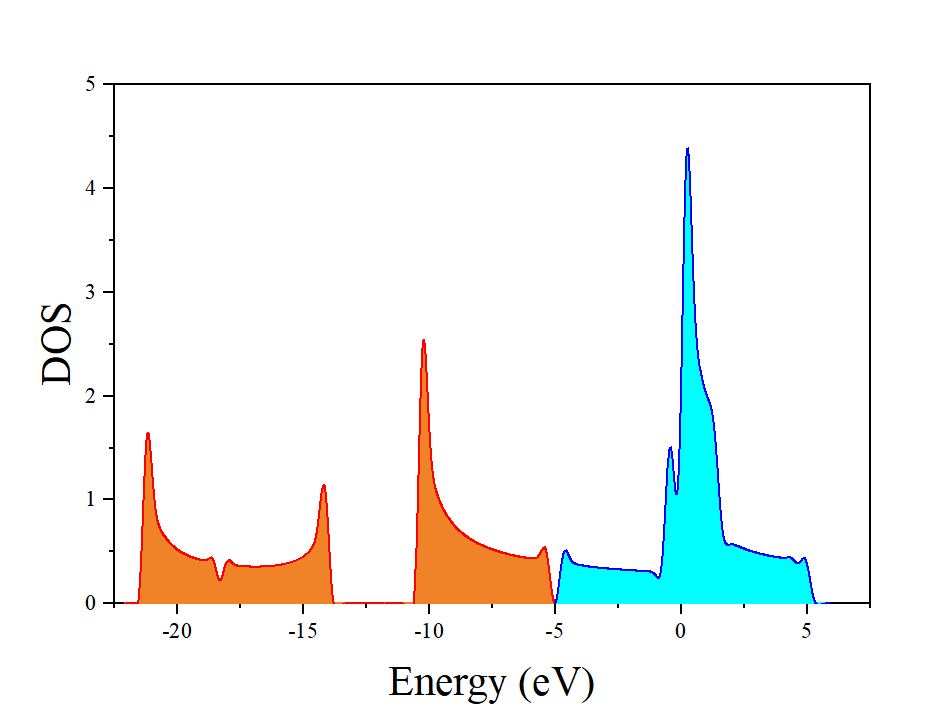
对计算出来的态密度采用OriginPro进行作图,(采用较密kpoint计算的结果)如下所示
采用 projwfc.x 对
projwfc.in &PROJWFC prefix = 'carbyne' outdir = './outdir' ngauss = 0 degauss= 0.01 DeltaE = 0.5 filpdos= 'carbyne.pdos' filproj= 'carbyne.proj' / projwfc.x <projwfc.in>projwfc.out
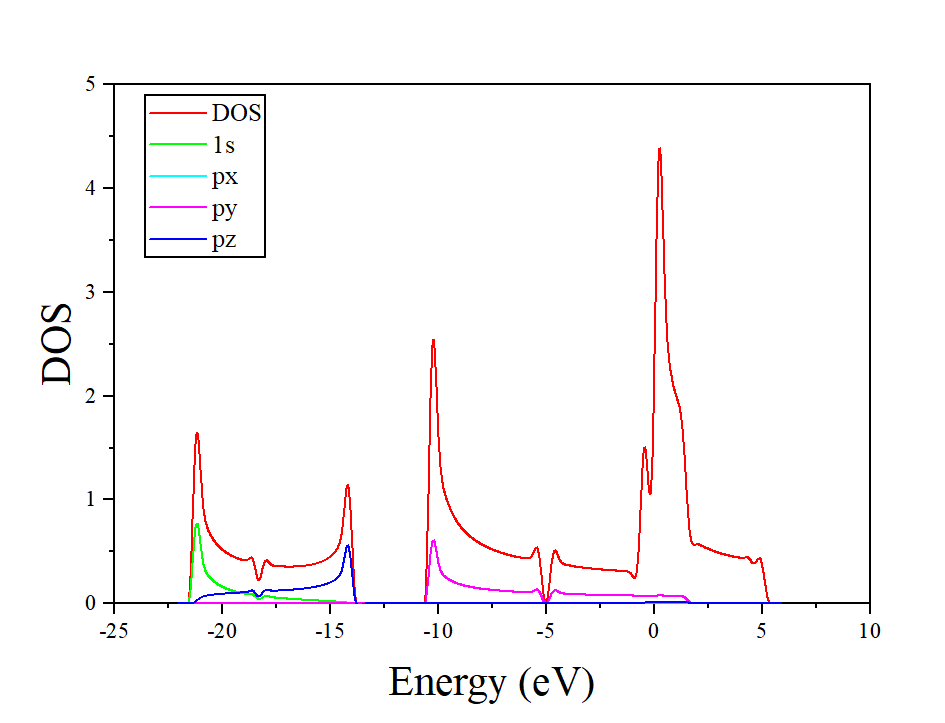
处理分波态密度得到的结果如下:
2.5 计算能带,修改[
calculation='bands'](https://www.quantum-espresso.o (https://www.quantum-espresso.org/Doc/INPUT_PW.html#K_POINTS)部分进行第一布cp -r scf band cd band cp scf.in pw-bands.in calculation = "bands" nbnd = 22 K_POINTS crystal_b 3 -0.5 0.0 0.0 50 0.0 0.0 0.0 50 0.5 0.0 0.0 1
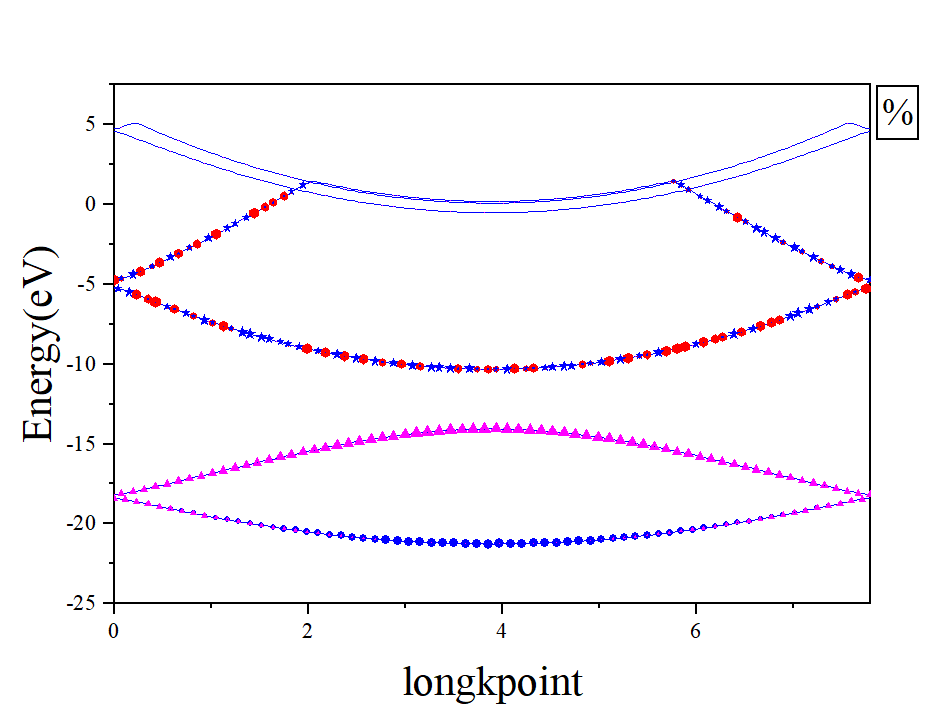
计算完成后使用[bands.x](http://www.quantum-espresso.org/Doc/INPUT_BANDS.htm x生成的文件bands.dat结合,画出各个能带的原子轨道投影,画图脚本如下:[/LVCSH 计算能带图如下:
2.6 根据fatband中的结果,进行wannier90计算
2.6.1 Scf计算
2.6.2 nscf计算
cp scf.in nscf.in并做如下修改 calculation = "nscf" nbnd = 16 !K_POINTS {automatic} ! 1 1 40 0 0 0 kmesh.pl 1 1 40 >>nscf.in pw.x < nscf.in > nscf.out
2.6.3 Wannier90计算
!System num_wann = 4 num_bands = 6 !Projection begin projections C : px;py end projections !Job Control exclude_bands : 1-2 !restart = !disentanglement dis_win_min = -25.0 dis_win_max = 7.0 dis_froz_min = -12.0 dis_froz_max = -0.6 dis_num_iter = 1000 dis_mix_ratio= 0.5 dis_conv_tol = 1.0E-10 dis_conv_window = 5 !Wannierise num_iter = 10000 conv_tol = 1.0E-10 conv_window = 10 guiding_centres = .true. ! SYSTEM begin unit_cell_cart Ang 20.000000 0.000000 0.000000 0.000000 20.000000 0.000000 0.000000 0.000000 2.565985410 end unit_cell_cart begin atoms_cart Ang C 10.0000000000 10.0000000000 -0.0019756118 C 10.0000000000 10.0000000000 1.2517014809 end atoms_cart ! KPOINTS
echo "mp_grid = 1 1 40">>${seedname}.win echo "begin kpoints">>${seedname}.win kmesh.pl 1 1 40 wannier >>${seedname}.win echo "end kpoints">>${seedname}.win
2.6.4 'wannier90.x -pp seedname'
2.6.5pw2wannier90.x < pw2wan.in > pw2wan.outpw2wan.in &inputpp outdir = './outdir' prefix = 'carbyne' seedname = 'carbyne' spin_component = 'none' write_mmn = .true. write_amn = .true. write_unk = .true. /
2.6.6
mpirun -np $NP wannier90.x seedname通过wannier90计算,找到进行wann2.7 采用DFPT进行phonon 2.7.1
cp -r scf phonon, 设置scf.in 输入文件.其中电子自洽收敛需要设置的高 同,K_POINTS的值可以设置的高一些,以使得phonon band计算出来的声子谱收敛。outdir = './' !nbnd = 22 conv_thr = 1.0e-9 K_POINTS {automatic} 40 1 1 0 0 0
2.7.2 进行phonon计算。 quantum-espresso.org/Doc/INPUT_PH.html#fildvscf)以输出自洽势对normal mode的
phonons calculation &inputph ! recover = .true. tr2_ph = 1.0d-16, prefix = 'carbyne', outdir = './' lraman = .true. ! epsil = .true. !use for insulators ldisp = .true. nq1 = 40 nq2 = 1 nq3 = 1 ! amass(1) = 0.0 fildyn = 'carbyne.dyn' fildvscf = 'dvscf' /
2.7.3 采用q2r.x进行力常数矩阵的傅里叶变换,得到实空间中的动力学矩阵,并添加
&input fildyn='carbyne.dyn', zasr='simple', flfrc='flfrc' /
2.7.4 使用matdyn.x 对实空间中力常数矩阵进行逆傅里叶变换,计算声子谱和声子态
&input asr='simple', !amass(1)=26.98, amass(2)=74.922, flfrc='flfrc', flfrq='phonon-freq', q_in_band_form=.true., / 2 0.0 0.0 0.0 100 0.5 0.0 0.0 1
使用plotband.x处理声子谱数据。plotband.in
phonon-freq 0 4000 phonon-freq.plot phonon-freq.ps 0.0 0.1 0.0
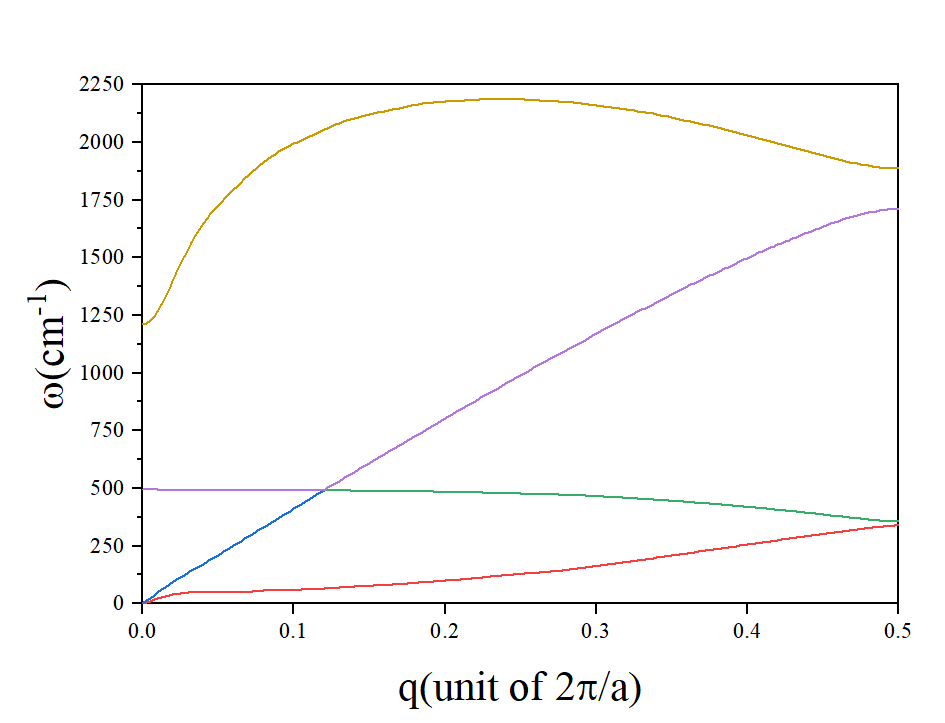
计算得到的声子谱如下图所示:
2.7.5 使用matdyn.x计算声子态密度. matdyn-dos.in
&input asr='simple', ! amass(1)=26.98, amass(2)=74.922, flfrc='flfrc', ! flfrq='flfrq', ! la2F=.true. dos=.true. fldos="phonon-dos" nk1=1,nk2=1,nk3=100 /2.7.6 使用pp.py(位于目录EPW/bin下)收集ph.x计算得到的fildvscf相关文件到s
2.8 EPW 计算电声耦合强度.计算
-
第一步:进入phonon目录进行scf自洽计算
-
第二步:ph.x进行DFPT计算(最费时间,需要注意设置参数
fildyn和fildvscf) 在phonon计算中可以使用 [-nimage N](http://www.quantum-espresso.org/Doc/ split automatically using the -nimage flag. See the phonon user guide for furth information.mpirun -np $NP -machinefile ${CURDIR}/nodelist ph.x -ni 6 -npool 28 <ph.i
-
第三步:使用pp.py收集ph.x计算得到的fildvscf相关文件到save文件夹
-
第四步:进入epw目录,先进行scf计算(或者将phonon目录中的内容拷贝过来), 计算时相同的参数设置和计算精度。
kmesh.pl 40 1 1 >>${prefix}.nscf.in -
第五步,设置 epw.in 文件,进行epw计算,设置[
prtgkk](https://docs.ep epw-code.org/doc/Inputs.html#fsthick)的设置,会影响打印出来的电声耦合矩阵元 epw.in如下:
epw calculation of carbyne &inputepw prefix = 'carbyne' outdir = './' amass(1)= 12.0107 dvscf_dir = '../phonon/save/' iverbosity = 0 elph = .true. ep_coupling = .true. ! epbwrite = .true. ! epbread = .false. epwwrite = .true. epwread = .false. ! etf_mem = 1 prtgkk = .true. ! ephwrite = .true. ! eig_read = .true. lifc = .true. asr_typ = 'crystal' wannierize = .true. nbndsub = 4 bands_skipped = 'exclude_bands = 1-2' num_iter = 10000 iprint = 2 ! dis_win_max = 12 ! dis_win_min = -25 ! dis_froz_min = -11 dis_froz_max = -0.2 proj(1) = 'C:py;pz' ! proj(2) = 'C:sp-1' write_wfn= .true. wannier_plot= .true. wdata(1)= 'bands_plot = .true.' wdata(2)= 'begin kpoint_path' wdata(3)= 'G 0.00 0.00 0.00 M 0.50 0.00 0.00' wdata(4)= 'end kpoint_path' wdata(5)= 'bands_plot_format = gnuplot' wdata(6)= 'conv_tol = 1.0e-10 ' wdata(7)= 'conv_window = 3 ' wdata(8)= 'dis_conv_tol = 1.0e-10 ' wdata(9)= 'dis_conv_window = 3 ' wdata(10)= 'dis_num_iter= 10000 ' wdata(11)= 'dis_mix_ratio= 0.5 ' wdata(12)= 'guiding_centres = .true.' wdata(13)= 'translate_home_cell : true' wdata(14)= 'translation_centre_frac : 0.0 0.0 0.0 ' elecselfen = .false. phonselfen = .false. a2f = .false. fsthick = 5.0 ! eV temps = 1 ! K degaussw = 0.005 ! eV nkf1 = 40 nkf2 = 1 nkf3 = 1 nqf1 = 40 nqf2 = 1 nqf3 = 1 nk1 = 40 nk2 = 1 nk3 = 1 nq1 = 40 nq2 = 1 nq3 = 1 /-
EPW声子谱和QE不一致 (参考简 这个问题一般是由于声子求和规则导致的,EPW中提供了读入实空间力常数来计算 ,然后再设置声子求和规则[
asr_typ = crystal](https://docs.epw-code. 且已经被命名为 ifc.q2r,对于包含SOC的情况,这个文件必须叫 **ifc.q2r. (https://forum.epw-code.org/index.php?f=3&t=137)&input fildyn='carbyne.dyn', zasr='simple', flfrc='ifc.q2r' /
-
第六步,使用第五步
epwwrite=.true.设置输出的wannier表象下的电声耦合文件
#!/bin/bash for i in $(seq 20 20 240) do cp epw.in epw${i}.in sed -i "s:epwwrite:epwwrite=.false. ! :g" epw${i}.in sed -i "s:epwread:epwread=.true. !:g" epw${i}.in sed -i "s:prtgkk:prtgkk=.true. !:g" epw${i}.in sed -i "s:wannierize:wannierize=.false. !:g" epw${i}.in sed -i "s:nkf1:nkf1=$i !:g" epw${i}.in sed -i "s:nqf1:nqf1=$i !:g" epw${i}.in cp qe-epw.bsub qe-epw${i}.bsub sed -i "2s:epw:epw${i}:g" qe-epw${i}.bsub sed -i "s:epw.in:epw${i}.in:g" qe-epw${i}.bsub sed -i "s:epw.out:epw${i}.out:g" qe-epw${i}.bsub bsub < qe-epw${i}.bsub sleep 10 done
- 上述脚本,有可能由于epwread文件读取冲突,导致不能正常计算,使用下列命令,
for i in $(seq 20 20 240);do echo epw$i.out;head epw$i.out;done
- In directory epw to calculate the electron-phonon coupling matrix using t kpoint and qpoint convergence.
-
-
构建目录,进行LVCSH.x的串行计算(只能单核计算,程序内的并行计算待开发),需
mkdir LVCSH-epw40 cp ./epw/epw40.out ./LVCSH-epw40 cd LVCSH-epw40 vi LVCSH.in LVCSH_complex.x &
calculation = "lvcsh" ! "lvcsh" or "plot" verbosity = "low" ! "low" or "high" l_dEa_dQ = .false. l_dEa2_dQ2 = .false. outdir = "./" methodsh = "FSSH" lit_gmnvkq = 0.0 ! in unit of meV lit_ephonon = 0.0 ! in unit of meV eps_acustic = 5.0 ! in unit of cm-1 lfeedback = .true. lehpairsh = .true. !lelecsh = .true. !lholesh = .true. !ieband_min = 3 !ieband_max = 4 !ihband_min = 1 !ihband_max = 2 !lsortpes = .false. !mix_thr = 0.8 epwoutname = "./epw.out" !nefre_sh = 40 !nhfre_sh = 40 nnode = 1 ncore = 1 naver = 2000 nsnap = 1000 nstep = 2 dt = 0.5 savedsnap = 25 ldecoherence = .true. Cdecoherence = 0.1 l_ph_quantum = .true. temp = 300 pre_nstep = 50000 pre_dt = 0.5 !gamma = 0.0 ! in unit of ps-1 ld_fric = 0.01 ! llaser = .true. efield_cart = 1.0 1.0 1.0 w_laser = 2.0 ! in unit of eV fwhm = 100 ! in unit of fs
-
构建目录使用手动方式进行LVCSH.x的并行计算(在不同的节点和核上进行不同轨迹的 make a directory LVCSH for lvcsh calculation。并在LVCSH目录下放入LVCSH.
mkdir LVCSH
3.1 make a lsf job script. Need to change the BUSB -q,-n,-R and MODULEPATH
lvcsh.bsub #!/bin/bash #BSUB -J JOB_NAME #BSUB -q QUEUE_NAME #BSUB -n ncore #BSUB -R "span[ptile=ncore]" #BSUB -o %J.out #BSUB -e %J.err #source ~/xh/.bashrc #export OMP_NUM_THREADS=1 #export MKL_NUM_THREADS=1 export MODULEPATH=$MODULEPATH:DIR_MODULEPATH module load lvcsh/version CURDIR=$PWD #Generate nodelist rm -f ${CURDIR}/nodelist >& /dev/null for i in `echo $LSB_HOSTS` do echo $i >> ${CURDIR}/nodelist done NP=`cat ${CURDIR}/nodelist |wc -l` for i in $(seq 1 1 $NP) do mkdir sample$i cp ./LVCSH.in ./sample$i/ cd ./sample$i LVCSH_complex.x & cd .. done wait
3.2 Use shell script mkepwdir.sh to build dir for diffrent kpoints and qpoi
nhfre_sh.mkepwdir.sh #!/bin/bash ncore=28 MODULEPATH="/share/home/zw/xiehua/opt/modules-4.7.1/modulefiles" lvcsh_version="0.6.6" QUEUE_NAME="privateq-zw" for i in $(seq 80 40 80) do mkdir epw$i mkdir epw$i/QEfiles cp ../epw/epw$i.out epw$i/QEfiles/ cp lvcsh.bsub epw$i sed -i "s/ncore/$ncore/g" epw$i/lvcsh.bsub sed -i "s:JOB_NAME:lvcsh-epw${i}-n0:g" epw$i/lvcsh.bsub sed -i "s:QUEUE_NAME:$QUEUE_NAME:g" epw$i/lvcsh.bsub sed -i "s:DIR_MODULEPATH:$MODULEPATH:g" epw$i/lvcsh.bsub sed -i "s:version:$lvcsh_version:g" epw$i/lvcsh.bsub cp job.sh epw$i cp LVCSH.in epw$i sed -i "s:./epw.out:../../QEfiles/epw$i.out:g" epw$i/LVCSH.in sed -i "s:ncore:ncore = $ncore !:g" epw$i/LVCSH.in cp lvcsh-test.bsub epw$i/lvcsh-plot.bsub sed -i "2s/JOB_NAME/lvcsh-epw$i-plot/g" epw$i/lvcsh-plot.bsub sed -i "s:QUEUE_NAME:$QUEUE_NAME:g" epw$i/lvcsh-plot.bsub sed -i "s:DIR_MODULEPATH:$MODULEPATH :g" epw$i/lvcsh-plot.bsub sed -i "s:version:$lvcsh_version:g" epw$i/lvcsh-plot.bsub cp LVCSH.in epw$i/QEfiles sed -i "s:verbosity:verbosity = "high" !:g" epw$i/QEfiles/LVCSH.in sed -i "s:./epw.out:./epw$i.out:g" epw$i/QEfiles/LVCSH.in sed -i "s:naver:naver = 10 !:g" epw$i/QEfiles/LVCSH.in sed -i "s:nsnap:nsnap = 2 !:g" epw$i/QEfiles/LVCSH.in sed -i "s:savedsnap:savedsnap = 2 !:g" epw$i/QEfiles/LVCSH.in cp lvcsh-test.bsub epw$i/QEfiles cd epw$i/QEfiles sed -i "2s/JOB_NAME/lvcsh-epw$i-test/g" lvcsh-test.bsub sed -i "s:QUEUE_NAME:$QUEUE_NAME:g" lvcsh-test.bsub sed -i "s:DIR_MODULEPATH:$MODULEPATH :g" lvcsh-test.bsub sed -i "s:version:$lvcsh_version:g" lvcsh-test.bsub bsub < lvcsh-test.bsub cd ../.. done
job.sh #!/bin/bash nnode=10 sed -i "s:nnode:nnode = $nnode !:g" LVCSH.in for i in $(seq 1 1 $nnode) do mkdir node$i cp ./lvcsh.bsub ./node$i/ sed -i "2s/n0/n$i/g" ./node$i/lvcsh.bsub cp ./LVCSH.in ./node$i/ cd ./node$i bsub < lvcsh.bsub cd .. done
LVCSH.in calculation = "lvcsh" ! "lvcsh" or "plot" verbosity = "low" ! "low" or "high" l_dEa_dQ = .false. l_dEa2_dQ2 = .false. outdir = "./" methodsh = "FSSH" lit_gmnvkq = 0.0 ! in unit of meV lit_ephonon = 0.0 ! in unit of meV eps_acustic = 5.0 ! in unit of cm-1 lfeedback = .true. lehpairsh = .true. !lelecsh = .true. !lholesh = .true. ieband_min = 9 ieband_max = 9 ihband_min = 8 ihband_max = 8 !lsortpes = .false. !mix_thr = 0.8 epwoutname = "./epw.out" !nefre_sh = 40 !nhfre_sh = 40 nnode = 1 ncore = 1 naver = 5000 nsnap = 1000 nstep = 2 dt = 0.5 savedsnap = 25 ldecoherence = .true. Cdecoherence = 0.1 l_ph_quantum = .true. temp = 300 pre_nstep = 5000 pre_dt = 0.5 !gamma = 0.0 ! in unit of ps-1 ld_fric = 0.01 ! llaser = .true. efield_cart = 1.0 1.0 1.0 w_laser = 2.0 ! in unit of eV fwhm = 100 ! in unit of fs
lvcsh-test.bsub #!/bin/bash #BSUB -J JOB_NAME #BSUB -q QUEUE_NAME #BSUB -n 1 #BSUB -o %J.out #BSUB -e %J.err export MODULEPATH=$MODULEPATH:DIR_MODULEPATH module load lvcsh/version LVCSH_complex.x
3.3 By look the initial adiabatic state in the QEfiles/LVCSH.out for differ calculation. Then, subscrib the job again.
bsub < lvcsh-test.bsub3.4. change the LVCSH.in file in the epw40, including the parameters for lv
ieband_min = 9 ieband_max = 9 ihband_min = 8 ihband_max = 8 epwoutname = "../../QEfiles/epw120.out" nefre_sh = 34 nhfre_sh = 34 nnode = 10 ncore = 28 naver = 100 nstep = 2 nsnap = 1000 dt = 0.5 savedsnap= 25
change the
nnodesin the job.sh bash script. Then './job.sh' to make3.5 After the setp 7, change the parameter
calculation = plotandepwout in all nodes and cores, then get a average results and writing in the dir